Вращаем и GOAT
Квантово-химические расчеты как хобби. Часть 2
(Это часть вторая, первую можно посмотреть по ссылке)
Немного вернусь к началу своего рассказа. В МТЦ СО РАН, где я работал и защитил кандидатскую, я, в основном, занимался свободнорадикальными реакциями в водном растворе. Это достаточно интересная тема, связанная с процессами деградации биомолекул типа белков и ДНК под действием различных факторов, например ультрафиолетового облучения.
Для анализа экспериментальных данных тех экспериментов, что тогда обсуждались в лаборатории, очень важны были сведения о структуре этих самых радикалов, особенно о том, где живет и как себя чувствует неспаренный электрон. Один из способов ответить на эти вопросы – это так называемые константы сверхтонкого взаимодействия (СТВ) ядер и неспаренного электрона в радикале. Именно эти константы я пытался рассчитывать при помощи квантово-химических расчетов.
Но там есть проблема, связанная со структурой органических молекул и радикалов. В них есть условно жесткие фрагменты, геометрия которых очень мало варьируется при комнатной температуре – ароматическое кольцо в производных бензола, например, а есть и очень подвижные относительно друг друга фрагменты – атомы углерода, связанные одинарными связями. Если для жестких фрагментов попытка попасть в правильную структуру при оптимизации геометрии частицы практически обречена на успех, то вот для подвижных фрагментов не все так радужно.
Я заметил это на своих результатах расчетов – пока меня интересовали жесткие структуры или фрагменты (например, в ароматических аминокислотах типа тирозина, триптофана или гистидина) – никаких особых проблем с описанием экспериментов не было. Но как только я отходил на шаг в сторону подвижных структур, пытался оценить то, что делают атомы рядом с жесткими фрагментами – никакого согласия с экспериментом не наблюдалось.
Но в декабре 2024 года, вооруженный новым умением сканировать геометрию, я решил попробовать подойти к этой проблеме чуть более систематично. Не просто перебирать случайно сгенерированные структуры в надежде на совпадение, а например, варьировать угол поворота вокруг связи и оптимизировать остальную геометрию частицы.
В качестве модельного объекта я взял катион-радикал этилбензола, смотри структуру ниже, сгенерировал в онлайн-редакторе MolView
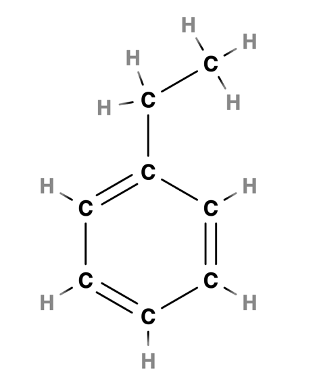
Здесь есть жесткий фрагмент – кольцо, и подвижный фрагмент – этильный заместитель в кольце.
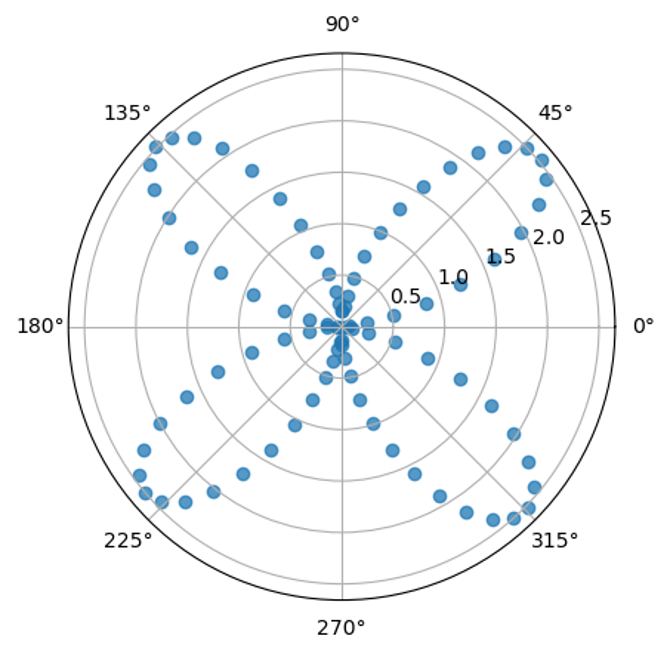
Сделал полный оборот этильного заместителя вокруг его связи с кольцом с шагом примерно в 4 градуса, записал на каждом шаге энергию частицы, потом построил график в полярных координатах, чем больше расстояние точки от центра, тем выше энергия системы, то есть тем менее выгодно это конкретное состояние.
Сделал полный оборот этильного заместителя вокруг его связи с кольцом с шагом примерно в 4 градуса, записал на каждом шаге энергию частицы, потом построил график в полярных координатах, чем больше расстояние точки от центра, тем выше энергия системы, то есть тем менее выгодно это конкретное состояние.
Потом я прогнал полученные геометрии через расчет констант СТВ (слава питону, что не вручную! 90 структур получилось). Эти константы я усреднил по Больцману, используя полученные на прошлом шаге энергии.
Почувствовав власть в своих руках, я попробовал нарисовать настоящую аминокислоту в редакторе, а именно фенилаланин, и повращать ту же связь для радикала. Здесь я натолкнулся на проблему, что молекула “сворачивается” определенным образом и не очень-то хочет из этого состояния выходить. На графике зависимости энергии от угла поворота это выглядит как резкие скачки:
Почувствовав власть в своих руках, я попробовал нарисовать настоящую аминокислоту в редакторе, а именно фенилаланин, и повращать ту же связь для радикала. Здесь я натолкнулся на проблему, что молекула “сворачивается” определенным образом и не очень-то хочет из этого состояния выходить. На графике зависимости энергии от угла поворота это выглядит как резкие скачки:
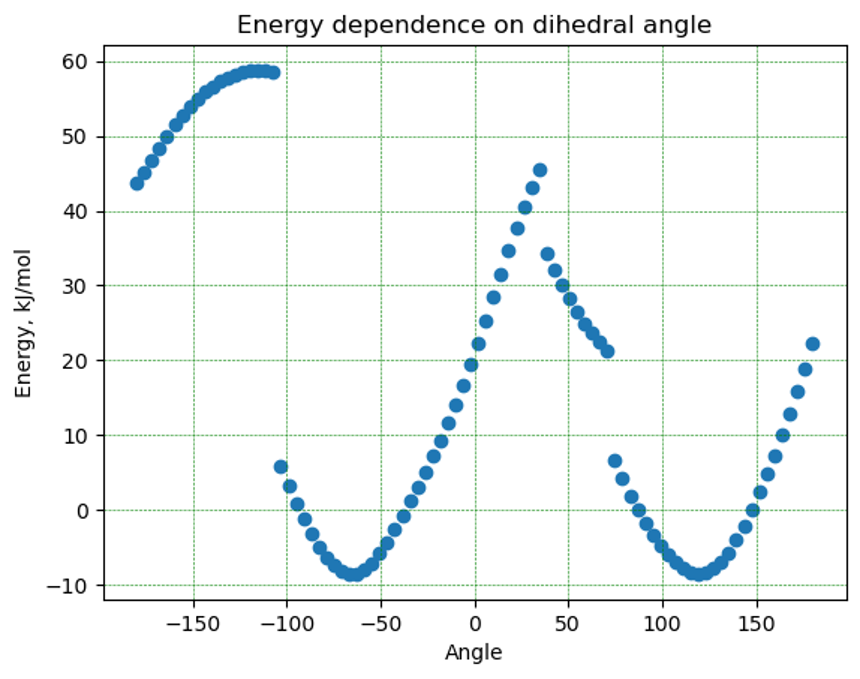
Плюс, зависимость не циклична, то есть при полном обороте молекулы я не попал в ту же точку по энергии, что свидетельствует о более сложном поведении системы.
Чуть приуныв, я пошел дальше читать мануал и туториалы к орке. Наткнулся на замечательную вещь, которая там называется GOAT (A Global Optimizer Algorithm). Если я правильно понимаю, то этот алгоритм позволяет имитировать нагрев молекулы с её последующим охлаждением, причем при охлаждении она с какой-то вероятностью попадает в различные локальные минимумы. Это полезно, во-первых, для адекватного поиска глобального минимума, и, во-вторых, исследования всего ансамбля различных вариантов структуры, которые способны взаимно превращаться друг в друга при комнатной температуре. Прогнал катион-радикал фенилаланина через эту процедуру, получил отрыв молекулы углекислого газа)) Понял, что оторвать электрон от этой конкретной молекулы тяжеловато, я перешел на те, что мы реально исследовали в лаборатории, а именно легко окисляемые ароматические аминокислоты: тирозин, триптофан, гистидин.
Начал с радикала тирозина, который может образовываться при фотоокислении тирозина в щелочной среде. GOAT XTB насчитал мне 37 структур с энергией в диапазоне 6 ккал/моль, этот порог говорит о том, что при комнатной температуре рассчитанные варианты еще имеют вклад в общий ансамбль, а те, что лежат выше – скорее не имеют. Рассчитал для каждой структуры константы СТВ, усреднил – и все равно не очень похоже на экспериментальные данные для бета-протонов. Заметил, что забыл оторвать протон от карбоксильной группы) Повторил расчет, получил такие данные
Как обычно, неплохо получились ароматические протоны (H2,3,5,6), но вот с бета-протонами что-то не совсем то, сильно разные для двух водородов и не очень попали в эксперимент. Чуда не случилось.
Здесь у меня есть некоторый вопрос для знатоков) Для GOAT я использовал метод XTB[2], жутко быстрый, но похоже, что не самый точный. Константы СТВ я рассчитывал методом B3LYP/EPR-II, пробовал брать энергии из выходных файлов этих расчетов, получалась совсем другая картина энергетического ландшафта системы. Возможно, стоит вообще каким-то третьим (каким?) методом посчитать не только энергии конформаций, но и частоты колебаний посмотреть на предмет мнимых частот, возможно дооптимизировать даже структуры… Оставлю себе на следующий заход эти рассуждения, похоже.
Чуть приуныв, я пошел дальше читать мануал и туториалы к орке. Наткнулся на замечательную вещь, которая там называется GOAT (A Global Optimizer Algorithm). Если я правильно понимаю, то этот алгоритм позволяет имитировать нагрев молекулы с её последующим охлаждением, причем при охлаждении она с какой-то вероятностью попадает в различные локальные минимумы. Это полезно, во-первых, для адекватного поиска глобального минимума, и, во-вторых, исследования всего ансамбля различных вариантов структуры, которые способны взаимно превращаться друг в друга при комнатной температуре. Прогнал катион-радикал фенилаланина через эту процедуру, получил отрыв молекулы углекислого газа)) Понял, что оторвать электрон от этой конкретной молекулы тяжеловато, я перешел на те, что мы реально исследовали в лаборатории, а именно легко окисляемые ароматические аминокислоты: тирозин, триптофан, гистидин.
Начал с радикала тирозина, который может образовываться при фотоокислении тирозина в щелочной среде. GOAT XTB насчитал мне 37 структур с энергией в диапазоне 6 ккал/моль, этот порог говорит о том, что при комнатной температуре рассчитанные варианты еще имеют вклад в общий ансамбль, а те, что лежат выше – скорее не имеют. Рассчитал для каждой структуры константы СТВ, усреднил – и все равно не очень похоже на экспериментальные данные для бета-протонов. Заметил, что забыл оторвать протон от карбоксильной группы) Повторил расчет, получил такие данные
Эксперимент[1], мТл | Расчет, мТл | |
Hbeta1 | 0,77 | 0,915 |
Hbeta2 | 0,77 | 0,394 |
Halpha | 0,04 | 0,014 |
H2 | 0,15 | 0,160 |
H3 | -0,615 | -0,578 |
H5 | -0,615 | -0,563 |
H6 | 0,15 | 0,157 |
Как обычно, неплохо получились ароматические протоны (H2,3,5,6), но вот с бета-протонами что-то не совсем то, сильно разные для двух водородов и не очень попали в эксперимент. Чуда не случилось.
Здесь у меня есть некоторый вопрос для знатоков) Для GOAT я использовал метод XTB[2], жутко быстрый, но похоже, что не самый точный. Константы СТВ я рассчитывал методом B3LYP/EPR-II, пробовал брать энергии из выходных файлов этих расчетов, получалась совсем другая картина энергетического ландшафта системы. Возможно, стоит вообще каким-то третьим (каким?) методом посчитать не только энергии конформаций, но и частоты колебаний посмотреть на предмет мнимых частот, возможно дооптимизировать даже структуры… Оставлю себе на следующий заход эти рассуждения, похоже.
- Tomkiewicz M., McAlpine R.D., Cocivera M. Photooxidation and decarboxylation of tyrosine studied by EPR and CIDNP techniques // Can. J. Chem. NRC Research Press Ottawa, Canada, 1972. Vol. 50, № 23. P. 3849–3856.
- Bannwarth C. et al. Extended tight-binding quantum chemistry methods // WIRES Comput. Molec. Sci. 2020. Vol. 11, № 2. P. e1493.